Box 1. Traces of a common origin
方框 1.共同起源的痕迹
Despite extensive gene exchanges, which generate diversity, and the absence of homology at the nucleotide and amino acid levels for most phage pairs, we observed a finite and relatively small number of different virion structures. This raises the question as to whether these structural similarities can be explained by divergent or convergent evolution. A divergent evolution would indicate that viruses share a common ancestor and have diverged beyond detectable sequence homology, while maintaining the basic architecture of their structural proteins. A convergent evolution would suggest that viruses share no common ancestors, but rather have converged toward a structure that is particularly optimal to build a virion. While both can lead to a single common trait, the accumulation of similar structural characteristics seems to point toward the divergent evolution hypothesis and the existence of a common ancestor.
尽管广泛的基因交换产生了多样性,而且大多数噬菌体对在核苷酸和氨基酸水平上没有同源性,但我们观察到的不同病毒结构数量有限且相对较少。这就提出了一个问题:这些结构上的相似性可以用分化进化还是趋同进化来解释。分化进化表明病毒有一个共同的祖先,其分化程度已超出了可检测到的序列同源性,但其结构蛋白的基本结构仍保持不变。趋同进化则表明病毒没有共同的祖先,而是趋向于一种特别适合构建病毒体的结构。虽然两者都可能导致单一的共同特征,但相似结构特征的积累似乎指向了分化进化假说和共同祖先的存在。
First, the Tectiviridae phage PRD1 MCP fold is highly similar to that of the archaeal virus STIV135 and the mammalian adenovirus34. The MCP is a trimeric protein made of two eight- stranded jelly rolls (-barrels). There are four different ways to fold such jelly rolls, but that one is only seen in these viruses136. Other features shared between PRD1 and adenovirus, include a linear dsDNA genome with inverted terminal repeats, the organization of the MCP on the capsid surface and the structure of spikes at the virion surface137. Other viruses are shown to have a PRD1- like structure, such as Tectiviridae infecting Gram-positive hosts (PRD1 infects Gram-negative hosts), Corticoviridae, eukaryotic and archaeal viruses138. This above suggests a common ancestor to PRD1-like viruses.
首先,Tectiviridae 噬菌体 PRD1 MCP 的折叠与古生病毒 STIV135 和哺乳动物腺病毒34 的折叠高度相似。MCP 是一种三聚体蛋白质,由两条八股果冻卷(-barrels)组成。这种果冻卷有四种不同的折叠方式,但只有这一种在这些病毒中出现136。PRD1 和腺病毒的其他共同特征包括:带有倒置末端重复序列的线性 dsDNA 基因组、荚膜表面的 MCP 组织以及病毒表面的尖峰结构137。其他病毒也有类似 PRD1 的结构,如感染革兰氏阳性宿主的 Tectiviridae(PRD1 感染革兰氏阴性宿主)、Corticoviridae、真核和古细菌病毒138。这表明类似 PRD1 的病毒有一个共同的祖先。
Second, a relationship also exists between tailed dsDNA phages, the archaeal virus HSTV- 154 and herpesviruses139. The MCP of these viruses has a common fold, called the HK97 fold. Several other structural similarities exist in HK97-like viruses, such as the presence of a portal on one vertex of the capsid and their capsid assembly pathways140. A third case of similarities involves Cystoviridae phage phi6 and phi8 with eukaryotic viruses belonging to Reoviridae (blue tongue virus, BTV) and Totiviridae141. These dsRNA viruses share a similar inner coat protein142 and have a segmented genome packaged in a double-shelled capsid137.
其次,有尾 dsDNA 噬菌体、古细菌病毒 HSTV- 154 和疱疹病毒 139 之间也存在关系。这些病毒的 MCP 有一个共同的折叠,称为 HK97 折叠。HK97 样病毒在结构上还有其他一些相似之处,如在噬菌体的一个顶点上有一个入口,以及它们的噬菌体组装途径140。囊病毒科噬菌体 phi6 和 phi8 与真核生物病毒 Reoviridae(蓝舌病毒,BTV)和 Totiviridae141 存在相似之处。这些 dsRNA 病毒具有相似的内衣壳蛋白142 ,基因组被分段包装在双壳囊壳中137。
Such structural resemblances between viruses infecting hosts spanning all three domains of life provide clues toward understanding the origin of viruses. Based on the previous examples of common ancestors, it has been proposed that viruses form polyphyletic lineages (PRD1-like, HK97-like and BTV-like) in contrast with the monophyletic origin of cellular life143,144.
病毒感染的宿主跨越生命的所有三个领域,它们之间的这种结构相似性为了解病毒的起源提供了线索。根据之前共同祖先的例子,有人提出病毒形成多单系(类 PRD1、类 HK97 和类 BTV),这与细胞生命的单系起源形成鲜明对比143,144。
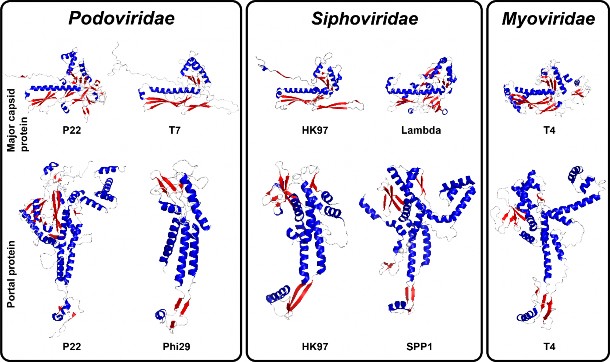
The tertiary structure of capsid or portal proteins protomers found in Podoviridae, Siphoviridae and Myoviridae. The HK97-like capsid protein structures were determined by X-ray diffraction for phages HK97 (PDB accession no. 10H6), T4 (PDB accession no. 1YUE) and lambda (PDB accession no. 3BQW) or cryo-EM for phages P22 (PDB accession no. 5UU5) and T7 (PDB accession no. 3J7W). The structure of the portal protein protomers was determined by X-ray diffraction for phages phi29 (PDB accession no. 1FOU), SPP1 (PDB accession no. 2JES), HK97 (PDB accession no. 3KDR) and P22 (PDB accession no. 3LJ4) or cryo-EM for phages T4 (PDB accession no. 3JA7). The coloring scheme used is based on secondary structures: red, β-strands; red, α-helices; grey, loops.
在 Podoviridae、Siphoviridae 和 Myoviridae 中发现的噬菌体外壳蛋白或门户蛋白原体的三级结构。噬菌体 HK97(PDB登录号:10H6)、T4(PDB登录号:1YUE)和 lambda(PDB登录号:3BQW)的类 HK97噬菌体噬菌体蛋白结构是通过 X 射线衍射法测定的,噬菌体 P22(PDB登录号:5UU5)和 T7(PDB登录号:3J7W)的类 HK97噬菌体噬菌体蛋白结构是通过冷冻电子显微镜法测定的。噬菌体 phi29(PDB登录号:1FOU)、SPP1(PDB登录号:2JES)、HK97(PDB登录号:3KDR)和 P22(PDB登录号:3LJ4)的入口蛋白原体的结构是通过 X 射线衍射测定的,噬菌体 T4(PDB登录号:3JA7)的入口蛋白原体的结构则是通过冷冻电镜测定的。所使用的着色方案基于二级结构:红色,β-链;红色,α-螺旋;灰色,环。
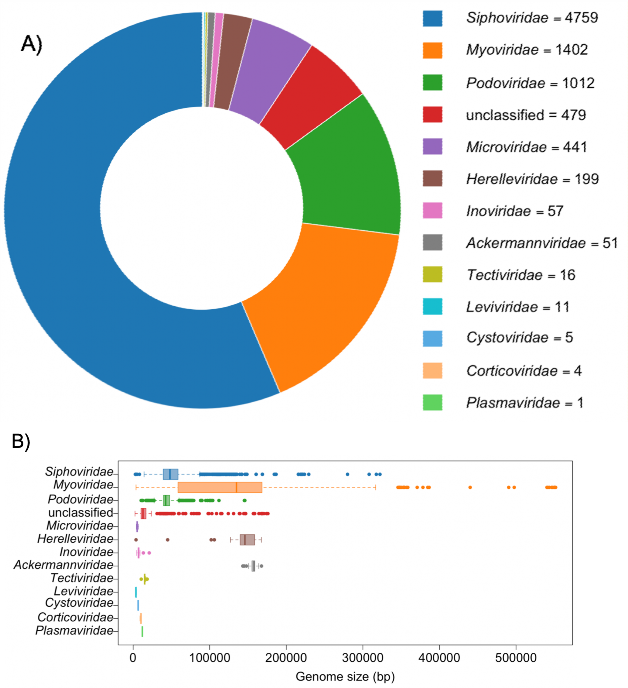
Fig. 2 Number of complete genomes (A) and genome size distribution (B) in each phage family as of September 2019 available in the NCBI Nucleotide database. The assignment of each phage to a family was done with the NCBI Taxonomy database. The unclassified group combines “unclassified Caudovirales”, “unclassified dsDNA phages” and “unclassified bacterial viruses”. This group is the fourth largest, emphasizing the increasing number of phages discovered through viral metagenomics for which no family can be assigned based on sequence information. Among the Caudovirales order, Herelleviridae and Ackermannviridae are the most homogenous families in terms of genome size. This is most likely because these two families were created after genomic analyses rather than morphological similarities.
图 2 截至 2019 年 9 月,NCBI 核苷酸数据库中每个噬菌体家族的完整基因组数量(A)和基因组大小分布(B)。每个噬菌体科的归属是通过 NCBI 分类数据库完成的。未分类组包括 “未分类的 Caudovirales”、”未分类的 dsDNA 噬菌体 “和 “未分类的细菌病毒”。该类是第四大类,强调了通过病毒元基因组学发现的噬菌体数量越来越多,而根据序列信息无法为这些噬菌体指定科属。在 Caudovirales 目中,Herelleviridae 和 Ackermannviridae 是基因组大小最单一的科。这很可能是因为这两个科是在进行基因组分析后创建的,而不是因为形态相似。
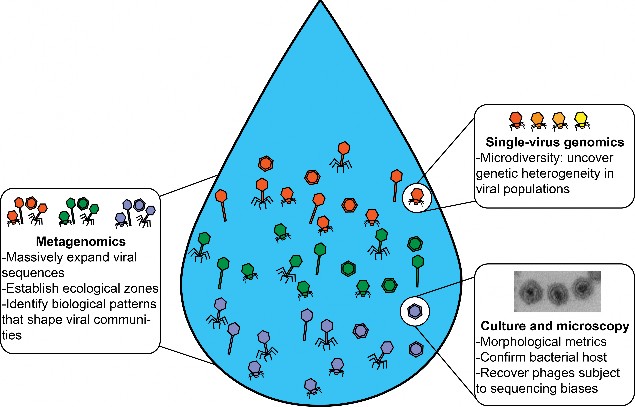
Fig. 3 Integrating metagenomics, single-virus genomics, culture, and microscopy to explore the viral dark matter. Several techniques have been developed to characterize phage diversity in biological communities, mostly from marine samples66. We focus here on techniques that do not require previous knowledge and that a priori can characterize the entire community. Metagenomics delivers the largest diversity of phages, with up to thousands of viral populations being identified11. Single-virus genomics enables sequencing of individual virions80. This helps to reveal phage populations with high levels of microdiversity (represented here by different shades of orange in the podovirus), which normally impede genome assembly in metagenomics pipelines. Culturing techniques combined with observations through a transmission electron microscope permit the discovery of phages otherwise subject to sequencing biases.
图 3 整合元基因组学、单病毒基因组学、培养和显微镜技术探索病毒暗物质。目前已开发出多种技术来描述生物群落中噬菌体的多样性,其中大部分来自海洋样本66 。在此,我们将重点关注那些不需要先前知识就能先验地描述整个群落特征的技术。元基因组学提供了噬菌体的最大多样性,可识别多达数千个病毒种群11。单个病毒基因组学可对单个病毒进行测序80 。这有助于揭示具有高度微观多样性的噬菌体种群(此处以荚膜病毒中不同深浅的橙色为代表),而微观多样性通常会阻碍元基因组学管道中的基因组组装。培养技术与透射电子显微镜的观察相结合,可以发现噬菌体,否则就会出现测序偏差。
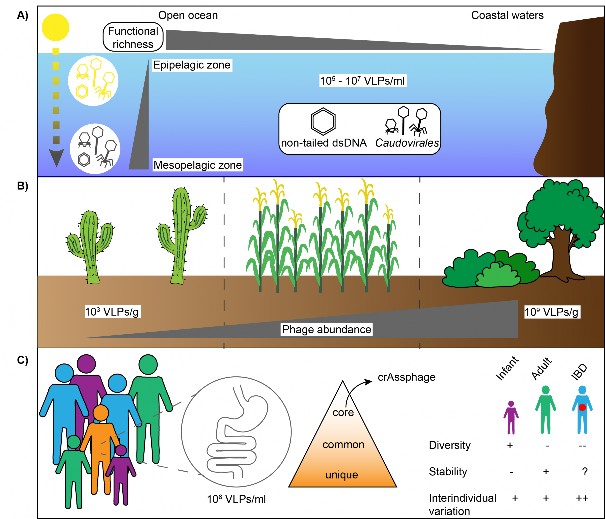
Fig. 4 Phage distribution and abundance in three ecosystems. A) Phages in the marine environment are extremely abundant with a virus to bacteria ratio often ranging from 1 to 100. qTEM of marine samples indicated that non-tailed phages are much more represented than tailed phages, which was also confirmed by metagenomic data6,145,146. Furthermore, phages from the mesopelagic zone were distinct from phages isolated from the epipelagic zone regarding gene content, life history traits and temporal persistence147. Similarly, functional richness was observed to decrease from deep to surface water and with distance from the shore for surface water only69. B) Phage abundance in the soil is also highly variable and correlates with biomes types, pH and bacterial abundance. Indeed, viral abundance is the lowest in hot desert, intermediate in agricultural soils and the highest in forest and wetland soils67. Viral abundance also positively correlates with bacterial abundance in the soil and negatively correlated with pH, with phage counts decreasing at higher pH. C) The phage community in the human gut is mainly composed of members of the Caudovirales and Microviridae and a large majority of these phages remain unclassified75,103,104. Phage composition is essentially unique to individuals, with global metagenomic analysis indicating that some phages are globally distributed75,76,103,104. The phage community is also stable during time, but rapid changes are observed in early life8. Changes in the diversity and composition of the human virome were also reported to be related to the gut health status, particularly in the case of inflammatory bowel disease (IBD)77,148.
图 4 噬菌体在三种生态系统中的分布和丰度。A) 海洋环境中的噬菌体极为丰富,病毒与细菌的比例通常在 1 到 100 之间。海洋样本的 qTEM 显示,非尾状噬菌体的数量远远多于尾状噬菌体,这一点也得到了元基因组数据的证实6,145,146。此外,在基因含量、生活史特征和时间持久性方面,来自中深海区的噬菌体与来自上深海区的噬菌体截然不同147。同样,从深层水到表层水,噬菌体的功能丰富度都在下降,而且仅表层水的噬菌体功能丰富度随距离海岸的远近而下降69。B) 土壤中的噬菌体丰度变化也很大,并与生物群落类型、pH 值和细菌丰度相关。事实上,炎热沙漠中的病毒丰度最低,农业土壤中的病毒丰度居中,森林和湿地土壤中的病毒丰度最高67。病毒丰度还与土壤中的细菌丰度呈正相关,与 pH 值呈负相关,pH 值越高,噬菌体数量越少。C) 人类肠道中的噬菌体群落主要由 Caudovirales 和 Microviridae 的成员组成,其中绝大多数噬菌体仍未分类75,103,104。噬菌体的组成基本上是个体独有的,全球元基因组分析表明,有些噬菌体是全球分布的75,76,103,104。噬菌体群落在一段时间内也是稳定的,但在生命早期会出现快速变化8。据报道,人类病毒组的多样性和组成的变化也与肠道健康状况有关,尤其是炎症性肠病(IBD)77,148。